With the development of life sciences research, more and more rare disease-related targets have become known to people, and therapies spawned by new strategies are no longer limited to conventional small molecules. Antibody therapy, peptide therapy, gene therapy, RNA therapy, cell therapy…a variety of new therapies are quietly emerging and thriving, and together built a vibrant ecology of rare disease therapies.
The therapies that this article focuses on include: peptide coupling therapy, oligonucleotide therapy, antibody therapy, cell therapy, gene therapy, small molecule therapy, etc.; covering diseases involving multiple myeloma, Merkel cell carcinoma, and systemic sclerosis disease-related interstitial lung disease, acute graft-versus-host disease, Fabry disease, retinitis pigmentosa, gangliosidosis, frontotemporal dementia, Krabbe disease, type A molybdenum cofactor deficiency, Alport syndrome chronic kidney disease, Waldenstrom’s macroglobulinemia, Prader-Willi syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, hereditary angioedema, etc.
Peptide Conjugated Therapy
- Pepaxt
On February 26, Oncopeptides announced that the US FDA has accelerated the approval of Pepaxto (melphalan flufenamide, also known as melflufen) to be marketed in combination with dexamethasone to treat adult patients with specific relapsed/refractory multiple myeloma (MM). Pepaxto is the first anti-cancer peptide conjugate drug approved by the FDA.
MM is a malignant blood cancer caused by abnormal proliferation of plasma cells in the bone marrow. Cancerous plasma cells can affect the production of normal blood cells, leading to decreased blood cell index, bone damage, and kidney damage. Although in the past ten years, the emergence of innovative therapies has significantly revolutionized the treatment of MM. However, many patients with MM will still relapse and develop resistance to existing therapies. Therefore, these patients with relapsed/refractory MM still need new treatment options.
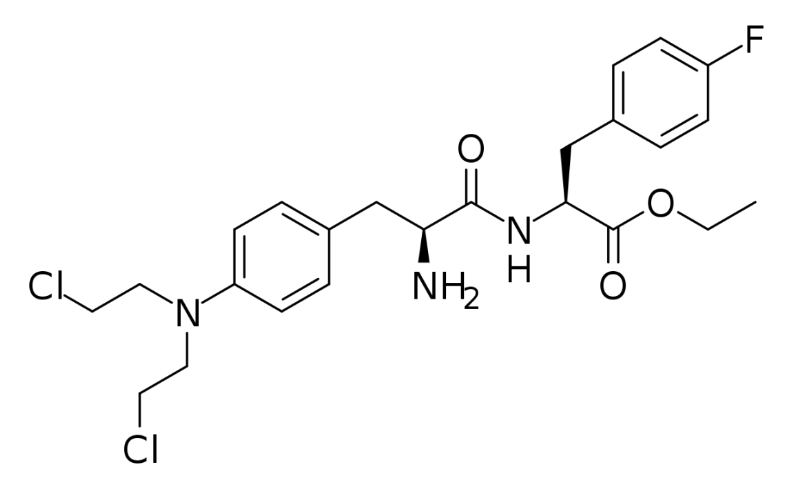
Figure 1: Pepaxto molecular structure.
Pepaxto is a “first-in-class” peptide-conjugated drug that conjugates alkylating agents with peptides that target aminopeptidase. Pepaxto can be quickly taken up by MM cells due to its lipophilicity. In the cell, it will be quickly hydrolyzed by peptidase, thereby releasing the hydrophilic alkylating agent. Aminopeptidase is overexpressed in tumor cells, especially in advanced cancers or tumors with high mutation complexes. In in vitro experiments, Pepaxto can increase the concentration of the alkylating agent in the cell, and its ability to kill MM cells is 50 times higher than that of the alkylating agent it carries.
Oligonucleotide Therapy
- Cavrotolimod (AST-008)
On March 3, Exicure announced that the US FDA granted its oligonucleotide therapy cavrotolimod (AST-008) orphan drug designation (ODD) for the treatment of Merkel cell carcinoma (MCC).
Cavrotolimod (AST-008) is a Toll-like receptor 9 agonists designed to activate the innate immune system and induce an effective anti-cancer immune response, especially when used in combination with checkpoint inhibitors.
Merkel cell carcinoma (MCC), also known as cutaneous neuroendocrine carcinoma, is a rare and highly aggressive tumor that tends to occur at the site of sun damage. MCC is derived from Merkel cells related to touch in superficial skin, and tumor cells contain neuroendocrine granules, so they have neuroendocrine cancer properties. MCC usually occurs on the face, neck, limbs, and other sun-irradiated areas. It is manifested as red skin bulges. The cause of the disease is not completely clear. About 80% of the cases are related to Merkel cell polyomavirus (MCV) infection. The continuous expression of T antigen may be one of its mechanisms.
Antibody Therapy
- Actemra (tocilizumab)
On March 5th, Genentech announced that the US FDA has approved the extended indications of the IL-6 receptor inhibitor Actemra (tocilizumab) for the alleviation of the rate of decline in lung function in adult patients with systemic sclerosis-related interstitial lung disease (SSc-ILD). Actemra is the first biological therapy approved by the FDA for the treatment of this disease.
SSc-ILD is a progressive lung disease in which lung function gradually declines and may be life-threatening. ILD is the leading cause of death in patients with scleroderma. Due to the gradual decline in lung function, the patient’s lungs cannot provide enough oxygen to the heart.
Actemra is a humanized IL-6 receptor antagonist, which has been approved for the treatment of patients with moderate to severe rheumatoid arthritis. This approval is based on the results of a randomized, double-blind, placebo-controlled Phase 3 clinical trial. In this study, compared with patients in the placebo group, patients treated with Actemra had a lower level of reduced forced vital capacity (FVC) at 48 weeks than in the control group. FVC decreased by 14 ml in the Actemra group and 255 ml in the control group.
- Neihulizumab(AbGn-168H)
On March 8, the US FDA granted AltruBio’s immune checkpoint regulator neihulizumab (AbGn-168H) fast track qualification for the treatment of steroid-refractory acute graft-versus-host disease (SR-aGVHD). Neihulizumab has previously obtained orphan drug designation from the US FDA for the treatment of aGVHD.
Graft versus host disease (GVHD) is an important cause of late non-relapse-related deaths after allogeneic hematopoietic stem cell transplantation (HSCT), and it seriously affects the quality of life of patients. The characteristics of GVHD are similar to the symptoms of autoimmune and other immune diseases, and its pathogenesis is not clear. It is currently believed to be mainly related to the negative selection of CD4+ T cells caused by thymus injury after transplantation, imbalance of cytokines in the body, imbalance of T cell ratio, and autoantibodies against the host synthesized by B cell, etc.
Neihulizumab is an immune checkpoint modulator targeting PSGL-1, which can promote the depletion of chronically activated T cells. This mechanism has been evaluated in four autoimmune and inflammatory diseases, and clinical proof of concept and safety has been verified in 170 patients. The Phase 1b multi-dose trial of SR-aGVHD is currently being studied. In addition, research on the first-line aGVHD is also underway.
Cell Therapy
- SIG-007
On March 5, Sigilon Therapeutics announced that the US FDA has granted its cell therapy product SIG-007 orphan drug designation for the treatment of Fabry disease, which has the potential to prolong the release of functional enzymes.
Fabry disease is a rare X-associated hereditary lysosomal storage disease caused by a defect in the activity of α-galactosidase A in the lysosome. The clinical manifestations of the disease are diverse, with burning pain in the extremities of the hands and feet as the common first symptom. In addition, patients will experience symptoms such as low sweating, no sweating, and heat intolerance in childhood, and even unable to live normally in severe cases. As the disease progresses, Fabry disease can also cause serious damage to the patient’s kidneys, heart, brain, nerves, and other organs. If effective treatment is not available, it may be life-threatening in severe cases. The current standard of care for Fabry disease requires intravenous administration, which may require a longer infusion time and may cause potential infusion-related reactions. Although these therapies have helped patients control the disease, long-term exposure has led to serious complications, including impaired kidney function.
SIG-007 consists of cells genetically modified with non-viral vectors to express human α-galactosidase A or AGAL. SIG-007 has the function of potentially sustaining and prolonging the release level of functional enzymes, thereby producing clinical benefits, and reducing the burden of treatment for patients.
- CiPC
On March 10, 2021, CiRC Biosciences announced that the US FDA has granted orphan drug designation (ODD) for chemically induced photoreceptor-like cells (CiPC) for the treatment of retinitis pigmentosa (RP).
RP is a group of hereditary blindness diseases characterized by progressive retinal photoreceptor-like cell apoptosis and pigment epithelial degeneration. Mutations in RP-related gene loci are the cause of the disease. There are various inheritance modes of RP, including autosomal dominant inheritance, autosomal recessive inheritance, and X chromosome-linked inheritance. The worldwide prevalence rate is 1/3,000 to 1/7,000.
CiRC Biosciences is currently advancing the preclinical development of CiPC, which is used for the treatment of advanced RP and the vision restoration of geographic atrophic age-related macular degeneration. A study conducted in a mouse model of RP showed that CiPC can lead to partial recovery of pupil reflex and visual function. The results of the study are published in the 2020 issue of Nature.
Gene Therapy
- PBGM01, PBFT02, PBKR03
On March 8, Passage Bio announced that the US FDA has granted it three fast-track qualifications for researching gene therapy, including PBGM01 for the treatment of GM1 gangliosidosis (GM1); PBFT02 for the treatment of GRN gene mutations Frontotemporal dementia (FTD-GRN); PBKR03 is used to treat Krabbe disease.
Small Molecule Therapy
- Nulibry (Fosdenopterin)
On February 27, the US FDA announced that it had approved Nulibry (fosdenopterin) developed by Origin Biosciences, a subsidiary of BridgeBio Pharma, to reduce the risk of death due to type A molybdenum cofactor deficiency (MoCD).
- Methyl bardoxolone
On March 1, Reata Pharmaceuticals announced that it had submitted a New Drug Application (NDA) for methyl bardoxolone to the US FDA for the treatment of chronic kidney disease (CKD) caused by Alport syndrome.
- Baiyueze (Zebutinib capsule)
On March 2, BeiGene announced that its self-developed BTK inhibitor Baiyueze (Zebutinib Capsule) has been approved by the Canadian Drug Administration for the treatment of patients with Waldenstrom’s macroglobulinemia (WM).
- Tesomet
On March 3, Saniona announced that the US FDA has granted Tesomet orphan drug designation for the treatment of Prader-Willi syndrome (PWS).
- Evrysdi(Risdiplam)
On March 9, Roche announced that the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) has issued a positive review opinion, recommending approval of Evrysdi (risdiplam) for marketing, treatment for more than 2 months, and clinical diagnosis of spinal cord Patients with all types of muscular atrophy (SMA) (type 1, type 2, type 3) or 5q-SMA with 1 to 4 copies of SMN2.
- AMX0035
On March 10, Amylyx Pharmaceuticals announced that it plans to submit a new drug application for the small molecule combination therapy AMX0035 in Canada for the treatment of amyotrophic lateral sclerosis (ALS). This application is expected to be finalized in the first half of 2021.
- Orladeyo(Berotralstat)
On March 11, BioCryst Pharmaceuticals announced that its once-a-day oral therapy Orladeyo (berotralstat) has been granted a French Temporary Authorization for Use (ATU) for the prevention of hereditary angioedema (HAE) in patients 12 years and older. attack.