Antiviral drugs
Antiviral drugs are a class of drugs used to treat viral infections. While most antiviral drugs target specific viruses, broad-spectrum antivirals are effective against multiple viruses. Most of the antiviral drugs available nowadays target HIV, herpes, hepatitis B and C viruses, and influenza A and B viruses. The coronavirus represented by COVID-19 has also become the focus of antiviral drug development.
Peptide and peptidomimetic antiviral drugs
The generic names of many antiviral drugs end in “vir” and the chemical structure of these antiviral drugs are in the form of peptidomimetics, such as atazanavir, lopinavir, nelfinavir, boceprevir, telaprevir, nematvir/ritonavir, not to mention antiviral drugs ending with “tide” including bulevirtide.
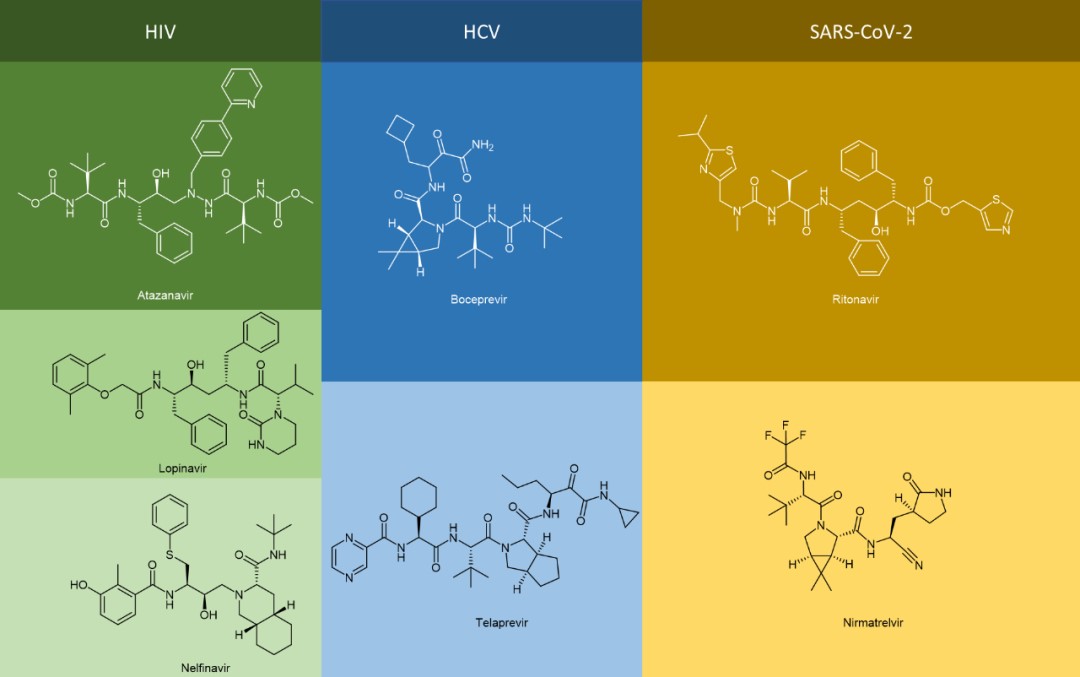
The use of peptidomimetics as a therapeutic agent has many advantages compared with peptide drugs, e.g., enhanced binding force, improved metabolic stability, and good bioavailability. It is an important means for the development of antiviral drugs of this kind to transform traditional peptide drugs into peptidomimetics through different strategies of chemical modification, to further improve receptor affinity and pharmacokinetic properties.
Common strategies for peptide modifications
Traditional peptide drugs are hampered by poor metabolic stability and low oral bioavailability, therefore, researchers use chemical modifications to enhance their stability and bioavailability. Chemical modifications of peptides generally include the following methods:
1. N-Terminal modifications/C-Terminal modifications
The minimum action of chemical modification is to block the N-terminal of a peptide with an acetyl group and modify the C-terminal into an amide. Such modifications can reduce the Nα-mediated degradation of DKP (diketopiperazine). This protective effect is particularly significant for peptides with smaller molecular weights. By the end of 2021, among the 62 marketed short peptides (consisting of 15 amino acid residues or less), 37 peptides are modified at their N-terminal by acylation, pyroglutamate, or cyclization, and only 9 linear peptides have free Nα groups. Terminal modifications have a significant effect on the secondary structure of the peptide because it deprives the molecule of its electrical charge. The change in the spatial structure of the peptide, in turn, can affect molecular stability.
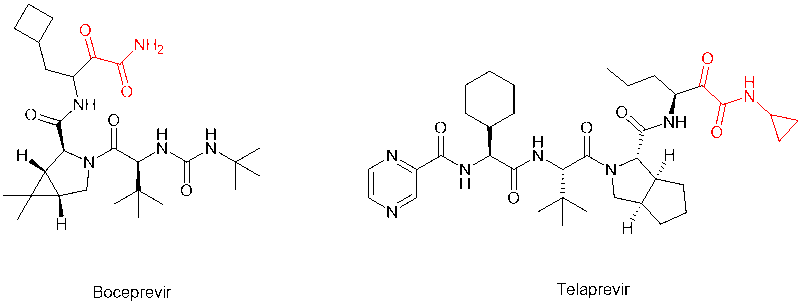
In addition to the two most common modifications above, the alpha-keto amide (alpha-keto-carbonyl amide) structure can be loaded on the C-terminal to introduce the keto-carbonyl group into the peptide molecule and transform it into a covalent peptide-like drug that can form a covalent bond with the receptor. For example, peptidomimetic drugs boceprevir and telaprevir, which target the serine protease of HCV virus, have alpha-keto amide in their C-terminus. The modification greatly improves the potency and pharmacokinetic properties of the parent compound.
Boric acid structures, another increasingly popular covalent drug, are also frequently introduced into peptide molecules. In a study, for example, scientists added boric acid structures to dipeptide inhibitors of Zika, West Nile, and dengue virus proteases, replacing the carboxylic acid or amide groups. As a result, the derived boric acid inhibitor had a thousandfold increase in affinity.
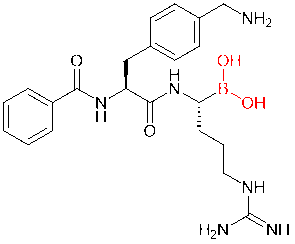
2. Bioisostereric replacement of amide
The skeleton of the peptide is composed of amide bonds, which is very important for the physical and chemical properties of the peptide. Replacing amide bonds by bioisostere is a vital strategy for researchers to move from peptide forward to peptidomimetics. Common amide electron isoplatelets include esters, thioamides, 1,2,3-triazoles, 1,2,3-triazoles, sulfonamides, phosphoramidites, hydroxy ethylene, hydroxy ethylamine, etc.
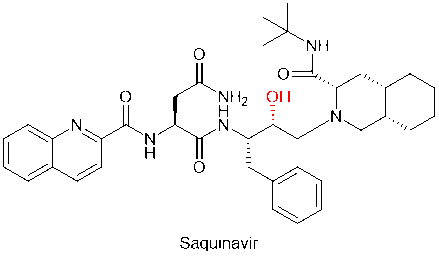
Practices of chemical modification methods that replace amide bonds by bioisostereric replacement are numerous in peptidomimetic drug development. For example, saquinavir, the first protease inhibitor against HIV, replaced the unstable amide bond in the peptide main chain with the hydroxymethyl structure, improving the metabolic stability of the drug.
Amino acid modifications are extremely extensive, including the substitution of non-proteinogenic amino acids for protein amino acids, such as D-amino acids for L-amino acids, thus improving the hydrolytic stability of the substrate. Researchers have a growing library of non-protein amino acids that can support different types of amino acid modification.
D-amino acids have even become regular residues of peptide drugs in the market. Among the peptide drugs approved in 2021 are voclosporin (D-Ala) and odevixibat (D-4-hydroxyphenylalanine), which contain D-amino acids. Furthermore, defelikefalin (korsuva, a pain-relieving opioid peptide for moderate to severe itching), approved by the FDA in 2021, uses a full D-amino acid (D-Phe, D-Leu, D-Lys) strategy.
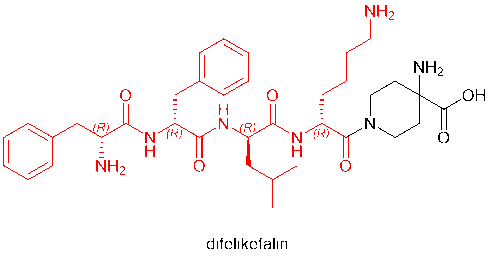
4. Cyclic Peptides
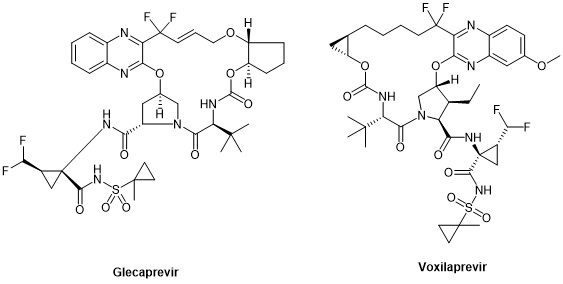
Peptide cyclization has always been an important chemical modification strategy for peptides. Since cyclosporin A, the first oral peptide preparation, was released in the 1980s, peptide cyclization has been regarded as one of the key ways to improve the stability, pharmacokinetic properties, and bioavailability of peptides. By 2021, 47% of the nearly 105 peptide therapeutic agents and diagnostic reagents approved by the FDA, EMA, and FMDA are cyclic peptides, of which 37% are single-cyclic peptides and 10% are polycyclic peptides. Cyclic peptides have also been found in anti-HCV drugs, especially in large cyclic reversible NS3/4A serine protease inhibitors, such as simeprevir, glecaprevir, and voxilaprevir.
R&D of Antiviral Peptides
1. Anti-SARS-CoV-2 virus peptidomimetic drugs
Perhaps the most high-profile example of the peptidomimetic antiviral therapy is Paxlovid, which helped stabilize the global COVID-19 outbreak caused by the SARS-CoV-2 virus. Paxlovid is an FDC (Fixed-dose combination) that contains two active substances, nirmatrelvir and ritonavir, which, as mentioned above, are peptidomimetics. Ritonavir was marketed as an anti-HIV drug but was quickly replaced by a new generation due to its potency and side effects. However, due to its high binding capacity with CYP3A4 enzyme and its inhibition of CYP3A4 mono plus oxidase (through different mechanisms, including (I) the formation of the metabolic intermediate complex (MIC) that closely bound to the heme group; (II) strong binding of unmodified ritonavir to heme iron; (III) heme destruction; (IV) covalent linking of active ritonavir intermediates to CYP3A4 deco-basing protein, etc.), ritonavir acts as a pharmacokinetic enhancer in Paxlovid.
Through its inhibition of CYP3A4, nirmatrelvir, the true antiviral active substance, is protected from enzymatic degradation of CYP3A4, increasing its half-life and drug exposure. The pharmacological action of nirmatrelvir is its inhibition of 3C-like protease (3CLPRO, also known as Mpro). Nirmatrelvir is therefore a Mpro protease inhibitor. Mpro is a cysteine protease whose cleavage of the S-glycoprotein on the spike protein of SARS-CoV-2 is critical to the biological process by which the virus fuses with host cells and infects them. Therefore, its inhibition is central to the pharmacological efficacy of Paxlovid.
Nirmatrelvir is a covalent peptidomimetic drug. Based on the non-covalent binding pocket to the receptor protein, the core building block of nirmatrelvir (the cyano group warhead), is the active electrophilic group on the covalent drug. It can interact with the sulfhydryl group of Cys145 residues on the Mpro protease to form thioimimate, which can bind the targeted receptor protein closely through covalently, and achieve high efficacy under the condition of a lower dose. It is important to note that Paxlovid is an oral antiviral based on the pharmacokinetic profile of the pharmacokinetic enhancer ritonavir and nirmatrelvir itself.

2. Anti-HCV peptidomimetic drugs
Anti-HCV (hepatitis C virus) peptide drugs are also one of the core tasks. Hepatitis C is a liver disease caused by the hepatitis C virus (HCV). Currently, treatment for HCV infection is based on direct antiviral drugs (DAAs) that target specific non-structural proteins and disrupt viral replication.
In 2011, the FDA approved first-generation DAAs, boceprevir and telaprevir, which target NS3/4A in patients with genotype 1 HCV infection and both contain alpha-keto amide in their C-terminus. These DAAs are not cheap to use in combination with pegylated interferon (peg-IFN) and ribavirin.
The second generation of DAAs, peptide-like drugs daclatasvir and sofosbuvir, which were introduced as NS5A and NS5B inhibitors, respectively, represent a breakthrough in HCV treatment, improving sustained viral response rate (SVR) in more patients.
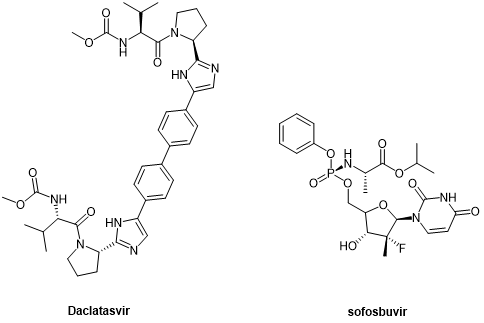
Glecaprevir and voxilaprevir, the third generation of DAAs developed for NS3/4A, target all genotypes, including genotype 3 that represents the milestone in HCV treatment, and are cyclic peptides.
3. Anti-HIV peptidomimetic drugs
Anti-HIV drugs can be divided into nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors, CCR5 antagonists, integrase strand transfer inhibitors (INSTIs), attachment inhibitors, post-attachment inhibitors, and many other types. Among protease inhibitors, many drugs are peptidomimetics.
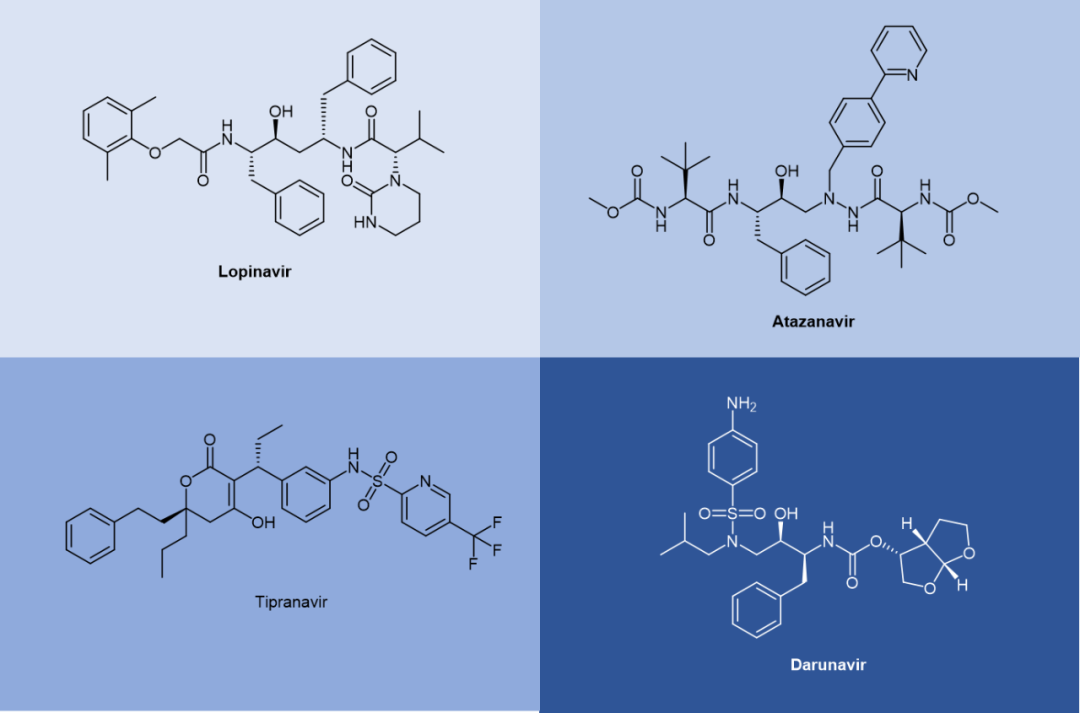
Saquinavir, a peptidomimetic drug approved by the FDA in 1995, was one of the first-generation protease inhibitors which were based on extranets such as hydroxy ethylene and hydroxy ethylamine electrons. The central hydroxyl group was bound to catalytic aspartic acid residues by simulating the transition state of the hydrolysis step. Ritonavir was also one of the first-generation anti-HIV protease inhibitors.
Lopinavir is a second-generation anti-HIV protease inhibitor developed by Abbott to improve the properties of ritonavir. Lopinavir cannot be administered alone and thus became the first compound preparation to be used in combination with ritonavir as a pharmacokinetic enhancer. Atazanavir was approved in 2003, and it became the first protease inhibitor to be effectively administered once a day. Its half-life is longer than that of previous protease inhibitors. Tipranavir was approved in 2005 and expanded for pediatric use in 2008. Because of many differences in its structure, it maintains effectiveness against certain strains of drug-resistant HIV and appears to have a higher genetic barrier, requiring many mutations to develop resistance. Darunavir, approved by the FDA in 2006, represents the latest anti-HIV protease inhibitor.
Summary
Peptidomimetics are powerful tools in pharmaceutical chemistry. They transform active peptides into complexes that combine the characteristics of peptides and small molecule drugs, significantly improving their pharmacological and pharmacokinetic properties over peptide drugs, and can be delivered orally on a large scale. The past decade witnessed a growing interest in antiviral peptides, and the emergence of Paxlovid demonstrated the tremendous value of antiviral peptides. With the deepening understanding of the pathogenic process of viruses, the combination of different antiviral drugs may become a reality, and the development of broad-spectrum antiviral drugs is also the research direction of scientists. It’s believed that the new generation of antiviral drugs ending with “vir” will continue to improve in efficacy, safety, resistance to drug resistance, delivery methods, and many other areas.
References
- Rossignol JF Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 2014, 110: 94–103.
- Rick Daniels; Leslie H. Nicoll. “Pharmacology – Nursing Management”. Contemporary Medical-Surgical Nursing. Cengage Learning, 2011. p. 397.
- Kausar S, et al. A review: Mechanism of action of antiviral drugs. International Journal of Immunopathology and Pharmacology. 202135: 20587384211002621.
- Yang, Y. Side Reactions in Peptide Synthesis. Elsevier. 2015, 22-28.
- King, T.A. et al. (2021) Photocatalytic methods for amino acid modification. Chem. Soc. Rev. 50, 39–57.
- Robello, M. et al. The Alpha Keto Amide Moiety as a Privileged Motif in Medicinal Chemistry: Current Insights and Emerging Opportunities. J. Med. Chem. 2021, 64, 7, 3508–3545.
- Nitshe, C. et al. Peptide–Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. J. Med. Chem. 2017, 60, 1, 511–516.
- Kumari, S. et al. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 21, 12290–12358.
- Ghosh, A.K. et al. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. 2016, J. Med. Chem. 59, 5172–5208.
- Allerton, C. Pain Therapeutics: Current and Future Treatment Paradigms. Royal Society of Chemistry. 2013, pp. 56
- Nageswara, D. et al. (2021) Discovery of quinoxaline-based P1–P3 macrocyclic NS3/4A protease inhibitors with potent activity against drug-resistant hepatitis C virus variants. J. Med. Chem. 64, 11972–11989.
- Loos, N, H. et al. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866.
- Kjellin, M. et al. The effect of the first-generation HCV-protease inhibitors boceprevir and telaprevir and the relation to baseline NS3 resistance mutations in genotype 1: experience from a small Swedish cohort. Ups. J. Med. Sci. 2018, 123 (1), 50−56.
- Wei, L. et al. Long-term outcomes in patients with chronic hepatitis C in the current era of direct-acting antiviral agents. Expert Rev. Anti Infect Ther. 2019, 17 (5), 311−325.
- Ghosh, A. K. et al. The FDA Approved HIV-1 Protease Inhibitors for Treatment of HIV/AIDS. In Burger’s Medicinal Chemistry and Drug Discovery, 7th ed.; Abraham, D. J., Rotella, D. P., Eds.; John Wiley & Sons: Hoboken, NJ, 2010; Vol. 7, pp 1−74.
- Bold, G.; et al. New aza-dipeptide analogs as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J. Med. Chem. 1998, 41, 3387−3401.
- Doyon, L.; et al. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 2005, 68, 27−35.