Cyclopeptides represent one of the most diverse architectures in current drug discovery efforts. Their small size, stability, and ease of synthesis provide an attractive scaffold for targeting and modulating some of the most challenging targets, including protein-protein interactions and those considered undruggable. Cyclopeptide libraries have been generated through various sophisticated screening techniques, including phage display, mRNA display, split-protein cyclization of peptides and proteins, and computer-based screening.
What are Cyclopeptides?
Cyclopeptides, with their small molecular weight (~500-3000 Da), high affinity, specificity, and ease of synthesis, remain one of the most attractive molecules in modern drug discovery. Compared to linear compounds, the conformational constraints imposed by cyclization make cyclopeptides highly resistant to proteolysis by endogenous proteases. Furthermore, their rigidity can improve their pharmacokinetic and pharmacodynamic properties and significantly impact their ability to passively penetrate cell membranes and reach intracellular targets. For instance, cyclosporins are small cyclopeptides derived from fungi and used to treat various diseases. They can transition between several different conformations, a unique feature contributing to their high passive membrane permeability.
In the past two decades, approximately 20 cyclopeptides have been approved by the FDA, confirming that cyclopeptides can exhibit properties similar to small molecules and antibody therapies. Due to the absence of established methods to reliably predict membrane permeability, most cyclopeptides have been developed for extracellular proteins, such as antibiotics like daptomycin. These antibacterial peptides target classical G-protein-coupled receptors (GPCRs).
Screening of Bioactive Cyclopeptides
With the advancement of modern technologies for synthesizing and screening cyclopeptide conformations with high affinity and specificity for target binding, the discovery of bioactive cyclopeptides has grown exponentially. Furthermore, advances in automated synthesis have simplified the fine-tuning of cyclopeptides, demonstrating ideal pharmacological properties. Designing, identifying, and selecting efficient, selective, and stable cyclic peptides from scratch have the potential to transform drug discovery from small molecules and antibodies to these novel molecules.
- Phage Display
George P. Smith discovered phage display, demonstrating that peptides could be displayed on the exterior of phage, viruses that infect bacteria, while encoding their genetic information internally. Subsequent refinements of this affinity-based in vitro technique led to the screening of libraries containing approximately 10^8 members for proteins and nucleic acid targets to identify high-affinity binding compounds. After selection and amplification, the top targets can be decoded and chemically synthesized or expressed in Escherichia coli for in vitro and in vivo analysis. In recent years, phage display has been used to develop cyclopeptide libraries containing unique macrocycle chemistry, introducing non-canonical amino acids and pharmacophores.
Ratmir Derda’s laboratory recently modified cyclization chemistry to install non-natural pharmacophores, targeting carbonic anhydrase (CA), an essential enzyme responsible for catalyzing the interconversion of carbon dioxide and water (Figure 1).
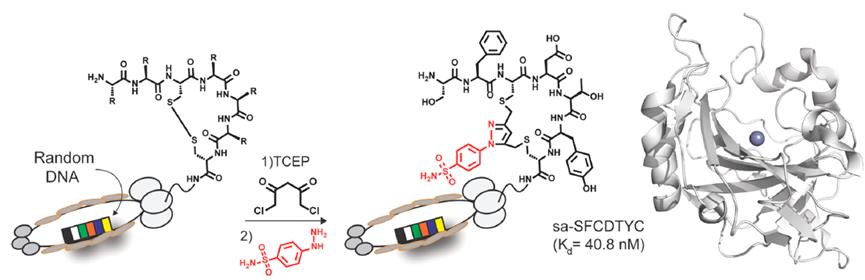
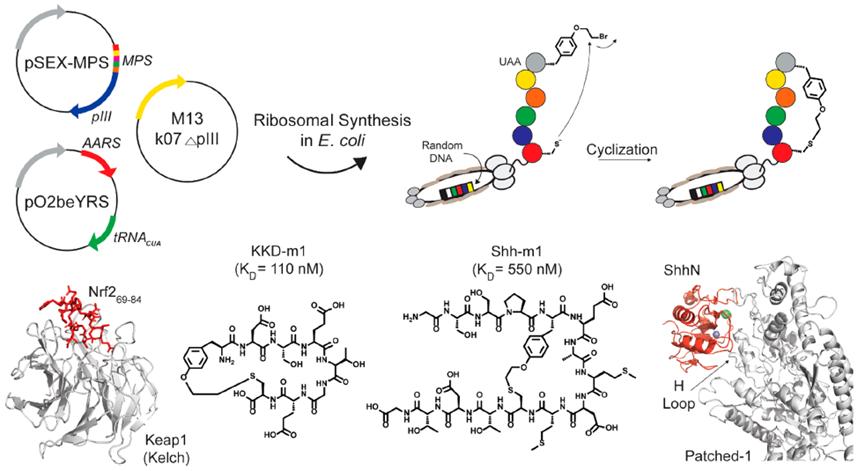
The Fasan laboratory has established a novel strategy by utilizing amber stop codon technology to introduce O-(2-bromoethyl)-tyrosine, enabling the cyclization of peptides on phage. This tyrosine can spontaneously react with proximal cysteine residues after translation to form irreducible thioether bonds (Figure 2).
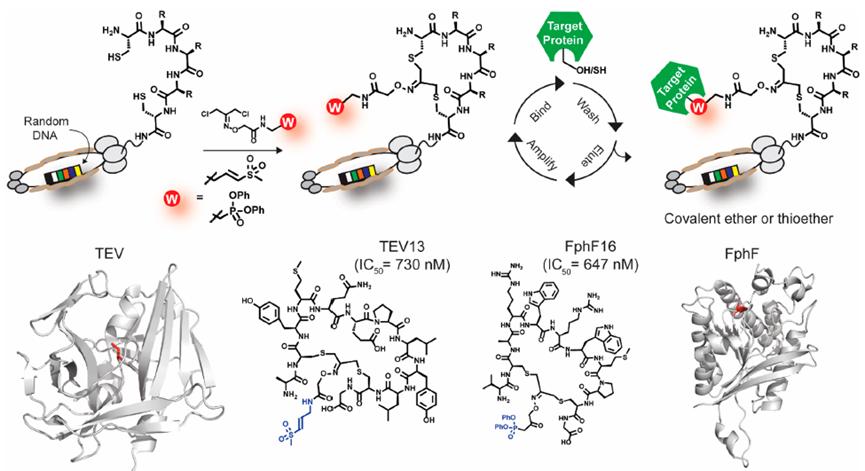
The Bogyo laboratory introduced an unbiased phage display screening strategy to identify irreversible covalent inhibitors of protein targets. By incorporating cysteine-reactive vinyl sulfone or serine-reactive diphenylphosphinyl ester heads linked to 1,3-dichloroacetone (DCA) into the cyclotide library, cyclopeptides can react with two cysteine residues in the library through DCA-modified heads (Figure 3).
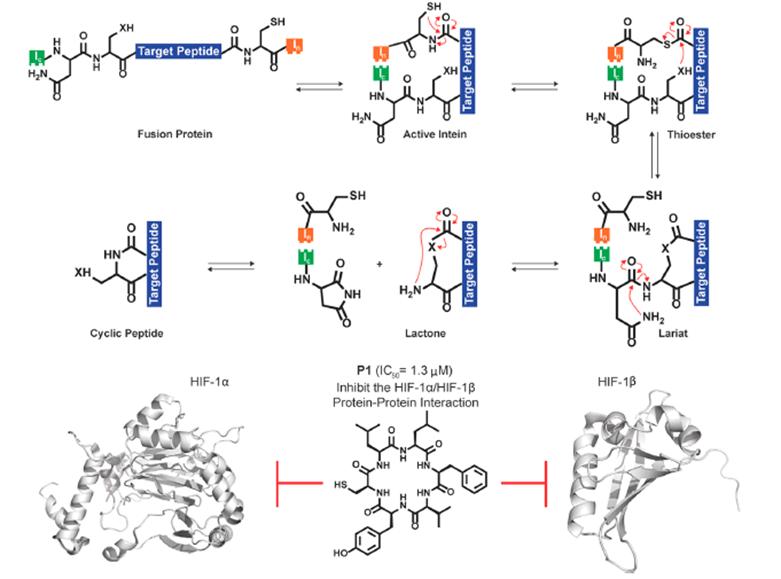
- Split-Intein Circular Ligation of Peptides and Proteins (SICLOPPS)
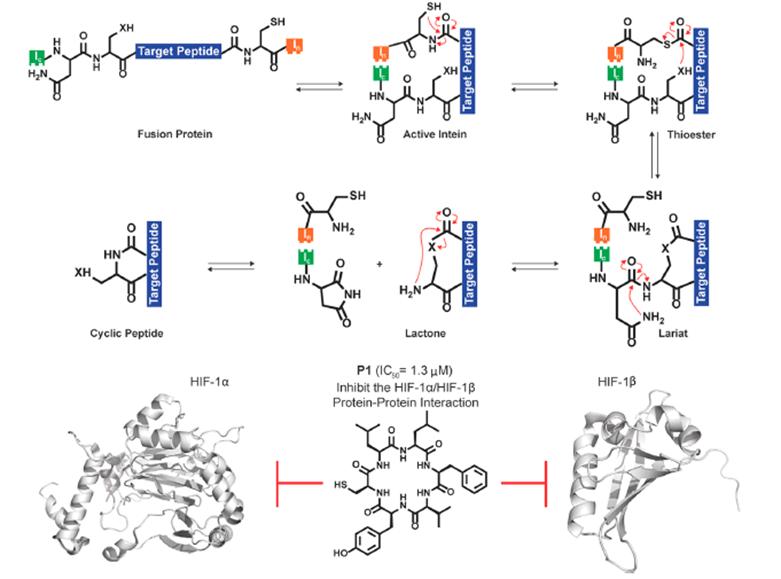
SICLOPPS is a novel method that screens cyclopeptides in live cells by utilizing the functional splicing of inteins, self-cleaving protein domains that excise their side-chain external protein domains. In this technique, the cyclopeptide of interest is inserted between split inteins in the form of IC-X-IN, where “X” represents the selected cyclopeptide sequence. After translation, the two intein fragments self-assemble, splicing out the cyclopeptide between them, releasing cyclopeptides with native amide bonds (Figure 4).
- mRNA Display
mRNA display is another effective in vitro screening method. Random nonstandard peptides integrated discovery (RaPID) is a mRNA display method promoted by Hiroaki Suga’s laboratory in 2006. This unique strategy utilizes flexible tRNA synthetases, allowing for the incorporation of various non-natural amino acids, including D-amino acids, N-methyl amino acids, and β-amino acids, significantly expanding the chemical space for screening. Moreover, genetic code reprogramming of N-terminal formylated Met can be replaced with amino acids, such as N-chloroacetyl-L-Tyr, which can spontaneously cyclize with proximal cysteine residues to generate cyclopeptide libraries. Recently, Suga’s lab used the RaPID system to identify effective and selective inhibitors of factor XIIa (FXIIa), a critical target for developing antithrombotic drugs (Figure 5).
- Structure-Guided Design
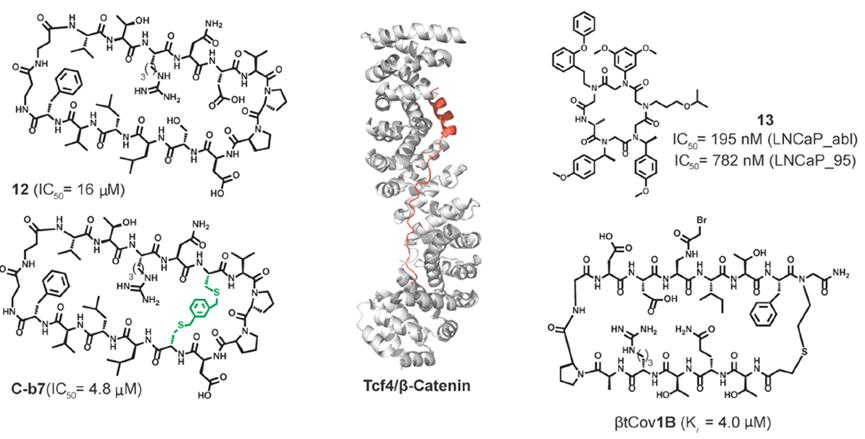
In recent research from Grossmann’s lab, cyclopeptide-designed inhibitors were developed to mimic the β-sheet characteristics of E-cadherin, targeting the interaction between Tcf4 and β-catenin. β-catenin is a transcriptional co-activator and a key component of the Wnt signaling pathway. Overactivation of this specific oncogenic target leads to various cancers, making it a target for therapeutic intervention. The binding constants of cyclopeptides were determined using in vitro fluorescence polarization (FP) assays. In this assay, competitive binding is detected through changes in fluorescence polarization when competitive ligands displace the fluorescently labeled Tcf4 ligand from β-catenin (Figure 6).
Challenges of Cyclopeptides
Although cyclopeptides are generally more stable than their linear counterparts, they can still degrade rapidly in vivo. One commonly used approach to enhance cyclopeptide proteolytic stability is introducing additional ring constraints to form bicyclic cyclopeptides. Another common approach is introducing non-natural amino acids, such as D-amino acids. As new screening technologies continue to emerge, such as the multiple analytical techniques of affinity selection-mass spectrometry (AS-MS), they may address the challenging issue of pushing cyclopeptides to the market.
Reference:
- Li, X., Craven, T. W., Levine, P. M., Cyclic Peptide Screening Methods for Preclinical Drug Discovery, J. Med. Chem., 2022, 65, 18, 11913-11926.