2024-07-09
GIP (Gastric Inhibitory Polypeptide or Glucose-dependent Insulinotropic Peptide) is a peptide hormone composed of 42 amino acids secreted by intestinal K cells, mainly produced in the duodenum and jejunum. GIP was initially found to have the function of inhibiting gastric acid secretion, but its more important role is as an insulin-stimulating hormone (Incretin), which promotes the release of insulin from pancreatic islet β cells in the presence of glucose, thereby effectively reducing postprandial blood glucose levels. In addition, GIP plays several important roles in fat metabolism, bone health, and other metabolic processes and diseases.
Studies have shown that GIP is not only expressed in the pancreas and stomach, but also widely distributed in multiple tissues such as adipose tissue, heart, adrenal cortex, and central nervous system. This suggests that GIP is not limited to the regulation of blood glucose, but also plays an important role in multiple physiological and pathological processes.
Regulates insulin secretion
The primary function of GIP is to stimulate insulin secretion by activating the GIP receptor (GIPR) on the surface of pancreatic islets β cells. The insulinotropic effect of GIP is particularly significant in hyperglycemic conditions, which is glucose-dependent and avoids the risk of hypoglycemia.
Regulation of fat metabolism
The role of GIP in fat metabolism is equally compelling. It promotes glucose uptake by fat cells and stimulates the activity of fatty acid synthase in fat cells, thereby promoting fat storage. At the same time, GIP enhances lipolysis by activating the expression of lipoprotein lipase (LPL). This two-way regulatory mechanism is closely related to the energy balance in the body.
Effect on bones
Studies have found that GIP has a role in bone health. By acting on osteoblasts, GIP can promote bone formation and inhibit the activity of osteoclasts. This property of GIP provides a new research direction for the treatment of bone-related diseases such as osteoporosis.
Other features
GIP is also involved in cell growth, differentiation, and metabolic regulation by activating MAP kinase and mTOR signaling pathways. Disorders of these functions can lead to pathological conditions such as insulin resistance, obesity, and even cancer. In addition, GIP may have indirect effects on energy intake and metabolism by regulating appetite through the nervous system.
GIP and GLP-1 (Glucagon-like Peptide-1) are the two main insulin-stimulating hormones, both secreted by intestinal cells, but their source cells and functional properties differ.
| Characteristic | GIP | GLP-1 |
|---|---|---|
| Source Cells | K cells (mainly in the duodenum and jejunum) | L-cells (mainly in the ileum and colon) |
| Target | Pancreatic islet β cells, adipose tissue, bones, etc | Pancreatic islet β cells, α cells, central nervous system |
| Key Features | Stimulates insulin secretion and regulates fat metabolism | Stimulates insulin secretion and inhibits glucagon |
| Metabolic Action | Promotes fat storage and breakdown | It mainly suppresses appetite and delays gastric emptying |
| Therapeutic Potential | Obesity, diabetes, osteoporosis, etc | Diabetes, obesity, metabolic syndrome, etc |
Although there is some overlap in the functions of the two, their specific roles in signaling pathways and metabolic diseases are complementary. Currently, therapeutic strategies combining GIP and GLP-1 receptor agonists are considered to have greater potential in the treatment of obesity and type 2 diabetes.
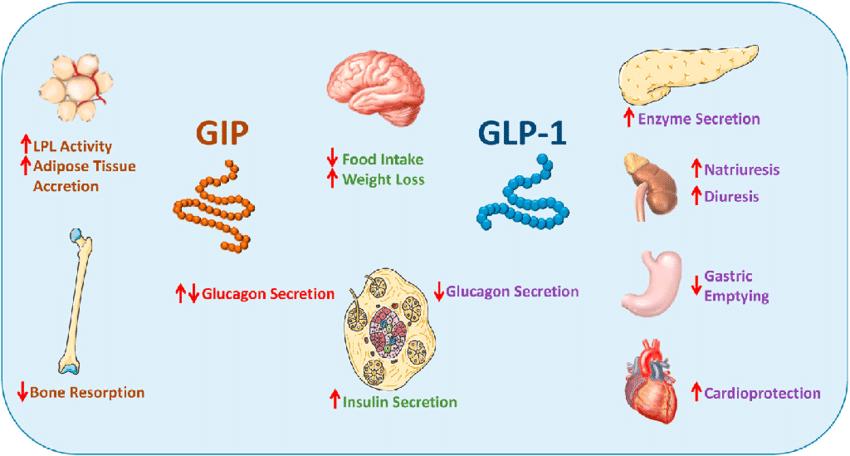 Fig.1 Metabolic actions of GLP-1 and GIP on key target tissues. (Baggio, Laurie L., and Daniel J. Drucker., 2021)
Fig.1 Metabolic actions of GLP-1 and GIP on key target tissues. (Baggio, Laurie L., and Daniel J. Drucker., 2021)
The biological function of GIP is achieved by binding to its receptor GIPR and activating a series of intracellular signaling pathways. GIPR is a receptor belonging to the G protein-coupled receptor family, which is widely expressed in the pancreas, adipose tissue, bones, and central nervous system.
Signaling pathways
GIP activates a variety of signaling pathways through GIPR, including:
cAMP/PKA pathway: Enhances insulin secretion and regulates cell metabolism and growth.
PI3K/AKT pathway: Involved in the regulation of fat metabolism and glucose metabolism.
MAPK and mTOR pathways: Associated with cell growth, differentiation, and increased risk of cancer.
Regulation of receptor expression
Studies have found that the expression level of GIPR is regulated by blood glucose, insulin, and inflammatory factors. In obese and type 2 diabetes patients, the function of GIPR may be inhibited, which is also one of the important mechanisms of metabolic dysregulation.
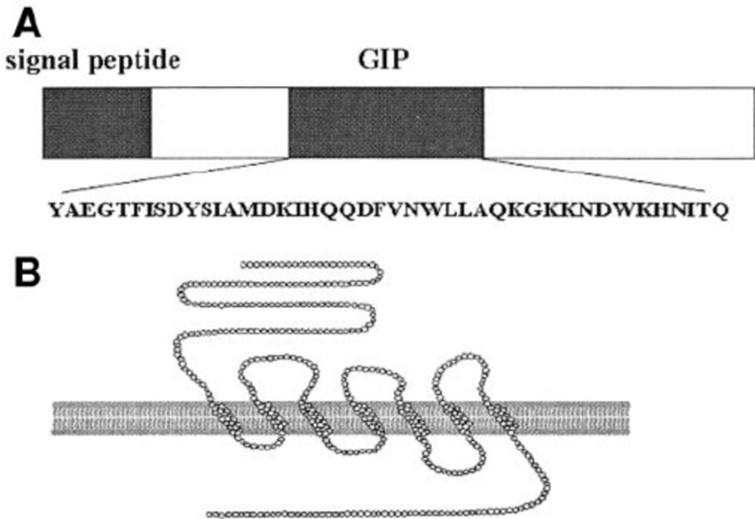 Fig.2 Structure of GIP and GIP receptor. (Yamada, Yuichiro, et al., 2006)
Fig.2 Structure of GIP and GIP receptor. (Yamada, Yuichiro, et al., 2006)
In recent years, with the in-depth research on the role of GIP in metabolic regulation, GIP receptor agonists have gradually become emerging drugs for the treatment of obesity and type 2 diabetes.
Development of dual agonists
GIP and GLP-1 dual receptor agonists have shown better efficacy compared to single agonists. By activating both GIPR and GLP-1R, these drugs are able to significantly reduce blood glucose levels, reduce body weight, and improve lipid metabolism. A representative of this class of drugs is Tirzepatide, which has shown excellent hypoglycemic and weight loss effects in clinical trials.
Potential side effects
Although GIP agonists have demonstrated significant therapeutic potential, their possible side effects are still of concern. For example, GIP may affect body fat distribution through fat metabolism and, in some cases, may exacerbate insulin resistance.
Table.1 Gastric inhibitory polypeptide at Creative Peptides.
| Product Name | M.W | Molecular Formula | Price |
|---|---|---|---|
| Gastric inhibitory polypeptide | Inquiry | ||
| [Tyr0] Gastric Inhibitory Peptide (23-42), human | 2584.9 | C119H182N34O31 | Inquiry |
| Gastric Inhibitory Polypeptide (6-30) amide (human) | 3010.47 | C139H209N35O38S | Inquiry |
| GIP (1 - 30), porcine, amide | 3551 | Inquiry | |
| Gastric Inhibitory Polypeptide (1-30), porcine | 3552.1 | C162H244N40O48S1 | Inquiry |
| Gastric Inhibitory Peptide (1-39), human | 4261.82 | C195H287N49O57S1 | Inquiry |
| GIP (3-42), human | 4759.4 | Inquiry | |
| (Pro3) Gastric Inhibitory Peptide (GIP), human | 4951.6 | C226H338N60O64S1 | Inquiry |
| GIP, porcine | 4975.6 | Inquiry | |
| Gastric Inhibitory Peptide (GIP), human | 4983.6 | C226H338N60O66S1 | Inquiry |
| GIP, mouse, rat | 5002.7 | Inquiry | |
| Acetyl Gastric Inhibitory Peptide (human) | 5025.6 | C226H338N60O64S1 | Inquiry |
| Gastric Inhibitory Polypeptide (porcine) | 4975.62 | Inquiry |
Role in type 2 diabetes
In healthy individuals, GIP effectively regulates blood sugar by promoting insulin secretion. However, in patients with type 2 diabetes, the response of pancreatic islet β cells to GIP is significantly weakened. This "GIP resistance" is one of the important reasons for poor postprandial blood sugar control in diabetic patients.
As a therapeutic target
Due to the extensive effects of GIP on fat metabolism and insulin secretion, the regulation of GIP signaling pathway may become an important direction in the treatment of diabetes. For example, GIPR antagonists can help with glycemic control by reducing fat storage and improving insulin sensitivity in some cases.
Treatments that combine GIP and GLP-1 are becoming a new trend. By modulating both signaling pathways, it can not only significantly improve blood sugar control, but also effectively reduce body weight and improve systemic metabolic health.
As an important incretin, GIP not only plays a central role in blood sugar regulation, but is also closely related to fat metabolism, bone health, and a variety of metabolic diseases. With the in-depth study of the GIP signaling pathway, its therapeutic potential in diabetes, obesity, and osteoporosis has been further confirmed. In particular, the combination of GIP and GLP-1 provides a new perspective and possibility for the management of metabolic diseases. In the future, drug development for GIP is expected to bring more breakthroughs to human health.
References