Since scientists discovered the first biologically active cyclic peptide (gramicidin S) in 1947, more and more cyclic peptides from marine organisms or microorganisms have attracted researchers' attention for their potential value in the field of medicinal chemistry. For example, cyclic depsipeptides are a kind of cyclic peptides that widely exist in sponges, cyanobacteria, and fungi, etc., and have shown various biological activities (such as anti-inflammatory, anti-HIV, and anti-tumor, etc.). These cyclic peptides from biological sources have inspired peptide researchers to convert defective natural linear peptides into cyclic peptides through chemical modification.
In the past, small molecule and antibody drugs have always occupied a broad therapeutic prospect, while peptides were considered "undruggable" because they target protein surfaces. The first two decades of the 21st century are the golden age of small molecule drugs. As far as the current FDA-approved drugs are concerned, small molecule drugs with a relative molecular weight less than 500 occupy an absolute advantage. Optimally designed small-molecule drugs can easily penetrate cell membranes, so they can target proteins localized in cell membranes. However, the nature of small-molecule drugs determines that there is usually a deep "pocket" near the active site of the target protein to facilitate the combination of small molecules and proteins. It is difficult to find suitable "pockets" for small molecule binding on 90% of the protein in the cell, so for small molecule drugs, only about 10% of the protein in the cell is druggable.
In recent years, due to the rapid development of peptide chemistry and the urgent need for protein-targeting drugs, it has been found that some peptide fragments with fewer amino acids have similar activities to proteins, and even have more prominent functions. In addition, they have good binding properties due to their relatively large volume and can interact with extended surfaces that traditional small molecules cannot target.
Compared with traditional drugs, peptide drugs have the following advantages:
(1) High efficiency: Significant activity can be exhibited at a lower dose or concentration;
(2) The molecule size is moderate: it is between small molecules and antibodies, which may have the characteristics of small molecules permeating membranes and antibodies mimicking protein functions;
(3) Low immunogenicity: Since the peptides are composed of amino acids, the design of the peptide sequence often uses a sequence homologous to humans, so the side effects are small;
(4) High synthesis efficiency: At present, peptide fragments with less than 50 amino acids can be quantitatively synthesized by solid-phase synthesis method.
Although peptide drug therapy has broad prospects, it still has the following shortcomings:
(1) Short half-life in vivo, easy to be degraded by various proteases, but lead to low oral utilization;
(2) Unstable binding to the target;
(3) It is not easy to enter the cell membrane to play a role.
In view of the above shortcomings, the hotspots of peptide drug research and development is mainly focused on maintaining all the activities of the original organism, and achieving the purpose of stabilizing the structure and enhancing the binding force of the target through external modification. As the best strategy to lock conformation and stabilize structure, cyclization has always been the focus of research.
More and more studies have found that cyclic peptides can significantly improve the activity of peptides. Compared with linear analogues, cyclic peptides have significantly improved target affinity, significantly increased stability against proteolytic enzymes, and, in some cases, easier entry into cells. At present, there are many discussions in the academic circle about the reason why cyclization can effectively improve the physicochemical properties of linear peptides. The classical explanation is that the conformation of linear peptides is relatively free and disordered, and cyclization gives linear peptides a certain degree of conformation locking and functional group preorganization, thus reducing the entropy loss caused by binding with the target.
Cyclic peptide drugs have the characteristics of good stability, high specificity, high affinity and low toxicity, which make them popular in the field of new drug development. There are also cyclic peptide drugs derived from endogenous somatostatin derivatives, such as Octreotide and its analogue Pasireotide, which have a longer half-life in vivo than somatostatin and can be used to treat acromegaly; There are also dual-target cyclopeptide inhibitors of MDM2 and MDMX, such as ALRN-6924, optimized by the cyclopeptide drug ATSP-7041, which can block p53 binding to MDM2 and MDMX and restore p53-mediated apoptosis of tumor cells.
At present, there are two main cyclization methods for peptides:
(1) Use functional side chains of natural amino acids for chemical modification and modification, such as lactam cyclic peptide, cysteine side chain sulfhydryl group for the synthesis of disulfide cyclic peptide and thioether cyclic peptide.
(2) The introduction of non-natural amino acids or groups with orthogonality to assist the synthesis of cyclic peptides, such as the cycloaddition reaction between alkynes and azides in click chemistry and the Michael addition reaction between thiols and alkenes.
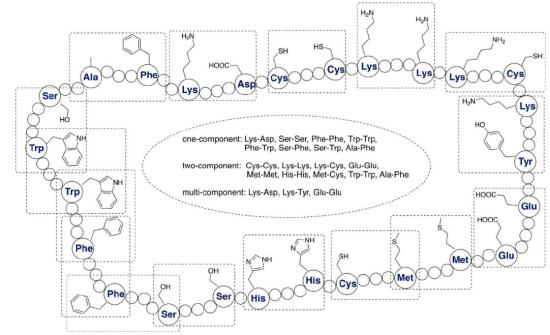 Common natural amino acid methods for constructing cyclic peptides
Common natural amino acid methods for constructing cyclic peptides
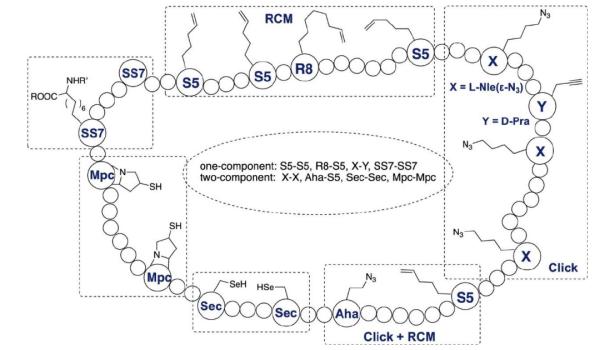 Construction of cyclic peptides based on non-natural amino acids
Construction of cyclic peptides based on non-natural amino acids
Since cyclization precursors carboxylic acids and primary amines are readily available in the peptide chains, lactam method is one of the earliest cyclization methods and can be used for the synthesis of head-to-tail macrocyclic peptides. The synthesis of lysine with aspartic acid or glutamic acid lactam into cyclic peptide is one of the most common methods in the synthesis of cyclic peptide. During the synthesis process, a variety of selective amino and carboxyl protective groups and deprotection mechanisms can be used to achieve lactam cyclization at specific sites, and even multiple lactam rings can be synthesized. Lactam cyclization can effectively improve the binding affinity of peptide drugs to receptors, and also show higher resistance to proteases. In this process, the reaction sites of lactam cyclization are mainly i,i+3; i,i+4 or i,i+7.
Disulfide bonds are an important part of the secondary or tertiary structure of natural proteins and are an essential part of protein function. Many natural proteins (such as insulin and neurotoxins) contain multiple pairs of disulfide bonds. At present, two disulfide-bonded cyclic peptides, ziconotide and linaclotide, have been approved by the FDA for the treatment of severe chronic pain, and the latter is used for the treatment of intestinal distress caused by constipation and chronic idiopathic constipation. Since the formation of disulfide bonds is reversible and usually exists in multiple pairs, the main challenge in the synthesis of such peptides is whether disulfide bridges can be formed at designated positions during the folding process. At present, the mainstream synthesis method is to introduce an orthogonal cysteine side chain protecting group, which is carried out by gradual removal and gradual oxidation. However, many pairs of disulfide bonds often have disadvantages such as isomerization, long steps and low yields in the synthesis.
Although the vast majority of cyclic peptide drugs currently used in clinical practice are derived from natural products, with the continuous optimization of research methods such as rational design and in vitro evolutionary screening, de novo design of cyclic peptides that target and bind to some receptors that natural peptides cannot bind has become a reality. With the wide application of high-throughput screening technology in the field of drug development and screening and human research on protein-protein interaction (PPI) networks, more and more cyclic peptide molecules are designed and screened for the treatment of some currently unresolved disease.
A bicyclic peptide is a peptide with two covalent rings that lock its structure, improving stability, target affinity, and resistance to enzymatic degradation compared with linear or monocyclic peptides.
Cyclization restricts conformational flexibility, preorganizes functional groups, and reduces entropy loss during binding, leading to stronger and more selective interactions with target proteins.
Common approaches include side-chain cyclization using cysteine or lysine residues, lactam formation, thioether bridges, and click chemistry with non-natural amino acids for precise ring construction.
Yes, their rigid and well-defined structures allow them to mimic protein surfaces and bind extended interfaces, making them valuable tools for probing or modulating protein-protein interactions in research.
Bicyclic peptides can be functionalized with fluorescent or affinity tags, serving as high-specificity probes for imaging, screening, or detecting specific biomolecular targets.
They provide high thermal and enzymatic stability, making them suitable for incorporation into biosensors, enzyme modulators, and other industrially relevant peptide-based systems.
By rational design, sequence variation, and strategic placement of cyclization points, researchers can enhance binding affinity, solubility, and resistance to degradation for specialized research applications.
Yes, high-throughput synthesis and combinatorial approaches allow generation of diverse bicyclic peptide libraries for screening target-specific binders, accelerating research and material discovery.
