2025-03-25
Research into drug discovery now centers on protein-protein interactions because they play essential roles in multiple signaling pathways. Creating small-molecule drugs that target Protein-protein interactions (PPIs) proves difficult because these interactions occur over extensive areas without clear binding sites. While monoclonal antibodies show promise for targeting PPIs they face limitations such as high expenses technical difficulties and inadequate intracellular penetration.
α-helices serve as crucial elements in protein-protein interactions and α-helical peptides represent a hopeful new avenue for developing pharmaceuticals. Short α-helical peptides fail to maintain their structure when dissolved in water which results in their instability and reduced bioavailability. Stapled peptides stand out as an effective approach because their side-chain cross-links reinforce the α-helix structure. The Verdine laboratory's research on stapled peptides showed their potential by modifying the BCL-2 protein which demonstrated intracellular activity and contested traditional views of "undruggable" targets.
Stapled peptides represent an innovative strategy for creating peptide-based medications that can be used to treat cancer and HIV and influence signaling pathways. The review presents the latest progress alongside classification methods and modification strategies to demonstrate how these innovations serve as potential next-generation therapeutics.
The carbon chain stapling bridge, particularly the alkene stapling bridge, is a classical method for synthesizing stapled peptides. In 1994, the Grubbs group reported the use of their discovered ring-closing metathesis (RCM) reaction for structural modification of peptides. This method involves synthesizing peptides first and then derivatizing serine residues at positions i and i+4 via side-chain allyl etherification, followed by RCM to form an α-helical peptide stabilized by a cyclic olefinic bridge. Later, Verdine and his team developed an alternative approach using non-natural amino acids with terminal alkene side chains to replace natural amino acid residues in peptides, forming cyclic olefinic bridges through RCM. This approach overcame the structural constraints of peptide side chains imposed by Grubbs' method, as well as the difficulties of liquid-phase cyclization synthesis. It also introduced the concept of "peptide stapling," which has since evolved into a widely used peptide modification strategy.
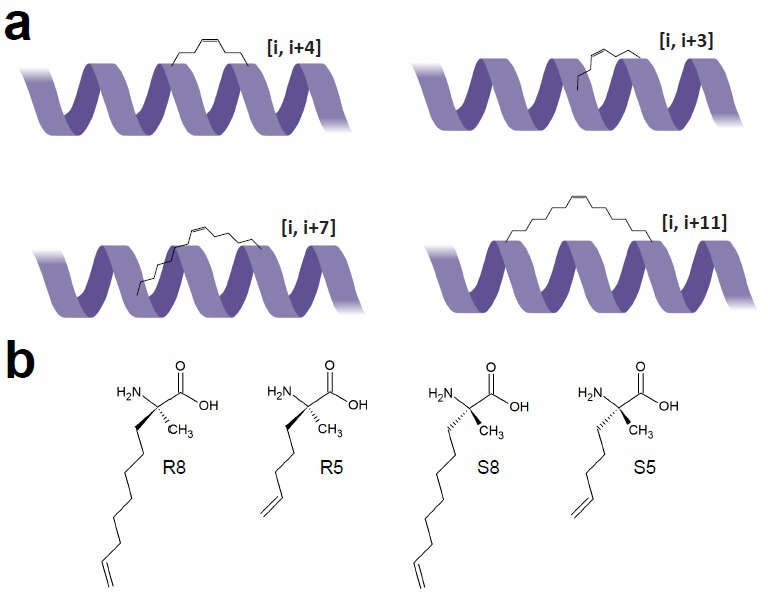 Fig.1 Structure of staple peptide1,2.
Fig.1 Structure of staple peptide1,2.
Table.1 Peptide modification service at Creative Peptides.
Cycloalkene-stapled peptides represent a classic form of stapled peptides and are one of the most extensively studied peptide modification strategies in terms of synthesis, structure, and activity. The most established method for constructing staples utilizes ruthenium catalysts to perform ring-closing metathesis. Studies have shown that RCM can be integrated with solid-phase peptide synthesis (SPPS), achieving complete conversion at room temperature within a few hours, outperforming many carbon-carbon coupling reactions that require prolonged heating and exhibit low conversion rates.
To construct the staple, two or more α-disubstituted non-natural amino acids must be incorporated into the peptide sequence as key building blocks. These non-natural amino acids typically feature an α-substituent with one methyl group and one alkene chain (e.g., pentenyl or octenyl). After assembling the target peptide chain via SPPS, the side chains (alkene groups) of two appropriately positioned non-natural amino acid residues undergo an intramolecular RCM reaction, eliminating an ethylene molecule to form a macrocyclic alkene bridge. Since this bridge locks the otherwise flexible helical peptide conformation like a staple, the resulting peptides are termed stapled peptides, with the bridge itself referred to as a staple. The α-disubstitution of the non-natural amino acids and the formation of the staple significantly enhance the stability of the α-helical conformation. Additionally, some amphiphilic helical peptides exhibit improved membrane permeability, while also overcoming the susceptibility of linear peptides to proteolytic degradation.
Table.2 Amino acid catalogue at Creative Peptides.
| Amino Acids & Derivatives | Glycosylated Amino Acids |
| Protected Amino Acids | Stable Isotope Labeled Amino Acids |
The key building blocks for stapled peptides are typically non-natural amino acids with α-alkene side chains. Current research focuses on chiral α-alkenyl non-natural amino acids with absolute configurations of either S or R, abbreviated as Sn or Rn (where n denotes the number of carbons in the alkene side chain). Additionally, α-dialkenyl-substituted amino acids, such as B5, are employed for stitched peptide modifications. The development of efficient, low-cost, and straightforward synthetic methods for these key building blocks is essential for stapled peptide synthesis.
Various research groups have established asymmetric synthetic methods for chiral amino acids. Notable approaches include:
Since B5 is an achiral amino acid, its synthesis is relatively simple. It can be obtained using N-diphenylmethylene glycine ester as the starting material, followed by two α-alkylation reactions, acid hydrolysis, and deprotection. The free Sn, Rn, and B5 intermediates are then converted into their Fmoc-protected forms (via Fmoc-OSu or Fmoc-Cl) for subsequent use in standard Fmoc-based SPPS.
Since the incorporation of non-natural amino acids alters the properties of the stapled peptide relative to the natural peptide, its activity in protein-protein interactions (PPIs) can also differ. Selecting optimal insertion sites for Sn, Rn, and B5 residues is critical for obtaining high-activity stapled peptides. Two primary strategies have been reported:
Stapled peptide Design: This approach involves analyzing the three-dimensional structure of the target protein and using computational modeling to optimize the α-helical peptide segment's conformation within PPIs. Non-natural amino acids are introduced at positions that do not participate in PPIs, ensuring that residues involved in binding remain intact. The staple is placed away from the interaction surface to avoid interference.
Combinatorial Screening: In this method, non-natural amino acids are systematically incorporated at different modifiable positions within the peptide sequence, generating a library of stapled peptides with various staple insertion sites. These peptides are then screened for biological activity to determine the optimal insertion site.
Although staple placement is generally avoided within the binding interface, in cases where hydrophobic amino acids (e.g., Val, Leu, Ile) mediate interactions, constructing an all-hydrocarbon staple on the interaction side can mimic these residues, allowing the staple to stabilize the helix and participate in binding. Speltz et al. further enhanced receptor binding by introducing methyl substituents into the alkene side chains of bridgehead amino acids.
Considering the structural features of α-helices, a complete turn consists of 3.6 amino acid residues. Within this structure, the side chains of residues at positions i, i+4, i+7, and i+11 align on the same side. Stapled peptide modifications utilize this alignment to perform RCM reactions on α-C side chains to form cycloalkene bridges.
The classical method for stabilizing one helical turn involves replacing the i and i+4 residues with two S5 residues, forming a staple. For stabilizing two helical turns, a staple is introduced at i and i+7, requiring longer side chains such as R8 + S5 or S8 + R5. Additionally, i and i+3 stapling is feasible but requires careful staple length selection: an 8-carbon staple requires R5 + S5, whereas a 6-carbon staple requires R5 + S3 or R3 + S5 to maintain the proper peptide conformation.
In 2014, the Verdine group introduced "stitched peptides," an innovative advancement of stapled peptide modifications. By incorporating the α-dipentenyl-substituted achiral amino acid B5 into the peptide sequence, along with Sn and Rn at adjacent positions, dual staples could be formed at i, i+4, i+8. i, i+4, i+11. or i, i+7, i+14. This stitching effect significantly enhances α-helical stability.
Currently, the S5 + S5 stapling strategy at i, i+4 is the most widely used approach. For short peptides, a single staple suffices to stabilize the α-helix, while longer peptides may require two or more independent staples (double-stapled peptides, distinct from stitched peptides). However, replacing S5 + S5 with R5 + R5 at i, i+4 is rarely reported, as it often reduces α-helicity and weakens cellular uptake.
The synthesis of stapled peptides follows standard SPPS procedures, with some modifications. Due to the steric hindrance of Sn or Rn residues, highly efficient coupling reagents such as PyAOP are required to ensure efficient peptide bond formation. The α-disubstituted amino acids cannot be monitored using the standard Kaiser test and must be confirmed via LC/MS after cleavage. To ensure that RCM occurs intramolecularly, the cyclization reaction is performed on solid support, leveraging the pseudo-dilution effect.
The synthesis process typically involves peptide chain assembly, RCM-mediated side-chain stapling, N-terminal modification, resin cleavage, and peptide purification, forming a well-established methodology.
Table.3 Peptide synthesis services at Creative Peptides.
The p53 gene is a crucial tumor suppressor gene closely related to DNA repair, cell differentiation, and cell cycle regulation. The deletion, mutation, or degradation of p53 by related proteasomes can lead to the formation of tumor cells. MDM2 and MDMX are the main inhibitors of p53, acting synergistically to suppress p53 activity through different signaling pathways. MDM2, an E3 ubiquitin ligase, binds to the transactivation domain of the p53 protein and mediates its ubiquitination, thereby reducing p53 stability. MDMX mainly inhibits p53 by binding to its transcriptional activation domain, thus suppressing its downstream gene transcription.
Studies have shown that during the interaction between p53 and MDM2 proteins, a 15-amino acid α-helix segment (LSQETFSDLWKLLPEN) in the N-terminal transactivation domain of p53 binds to the hydrophobic cleft of MDM2 and regulates their interaction, with Phe19, Trp23, and Leu26 being essential for MDM2 binding. Verdine's research group utilized an all-hydrocarbon side-chain stapling strategy to optimize these interactions. By screening the incorporation positions of alkenyl amino acids, modifying acidic and basic residues to adjust net peptide charge for improved membrane permeability, they synthesized and identified a stapled peptide, SAH-p53-8 (Ac-QSQQTF[R8NLWRLLS5]-QN-NH2). The modified non-natural alkenyl amino acids are indicated in bold, and the sequence within the stapled macrocycle is enclosed in brackets for precise representation. SAH-p53-8 exhibits 85% α-helicity and a dissociation constant (KD) of 55 nmol·L-1, enabling membrane penetration and inducing tumor cell apoptosis.
Subsequent studies revealed that SAH-p53-8 effectively inhibits MDMX with 25-fold higher affinity than MDM2, making it a promising dual-target tumor inhibitor for drug development. Further optimization using phage display mutation screening of the SAH-p53-8 lead sequence yielded a new peptide, Ac-LTFEHYWAQLTS-NH2. By replacing Glu4 and Thr11 with R8 and S5 to form a stapled structure, substituting Asn5 and Leu10, and optimizing the C-terminal structure, they developed the stapled peptide candidate ATSP-7041 (Ac-LTF[R8EYWAQCbaS5] SAANH2), which inhibits p53 binding to MDM2 and MDMX with inhibition constants (Ki) of 0.9 nmol·L-1 and 6.8 nmol·L-1, respectively. ATSP-7041 demonstrated significant tumor suppression both in vitro and in vivo with favorable pharmacokinetics. Further optimization led to the development of ALRN-6924, which has entered Phase II clinical trials (FDA Clinical Trial ID: NCT02264613) and is expected to become the first approved stapled peptide therapeutic.
The apoptosis-related BCL-2 protein family consists of two major classes: anti-apoptotic proteins such as BCL-2, BCL-XL, BCL-1, and MCL-1, and pro-apoptotic proteins including BAX, BCL-XS, BAK, and BID. Research has shown that the amphipathic α-helical segment in the BH3 domain of BCL-2 can bind to the hydrophobic region formed by BH1, BH2, and BH3, thereby promoting apoptosis.
Walensky and colleagues designed and synthesized a series of stapled peptides (SAHBs) by mimicking the α-helical BH3 segment of BID (EDIIRNIARHLAQVGDSNLDRSIW). Among them, the stapled peptide SAHBA (EDIIRNIARHLA[S5VGDS5] NLDRSIW) exhibited promising transmembrane capability, binding affinity, and apoptotic activity. SAHBA improved the KD value from 269 nmol·L-1 (unstapled peptide) to 38.8 nmol·L-1 and enhanced α-helicity to 87.5%. Additionally, SAHBA increased the serum half-life from 3.1 hours to 29.4 hours.
Subsequently, Stewart and colleagues screened a series of α-helical segments from the BCL-2 family and modified them into stapled peptides. They discovered that the BH3 helical segment from MCL-1 (KALETLRRVGDGVQRNHETAF) functions as a selective MCL-1 inhibitor. The modified stapled peptide MCL-1 SAHBD (KALETLRRVGDGV [S5RNHS5] TAF) exhibited an optimized KD value of 10±3 nmol·L-1, significantly improved from the original 245±29 nmol·L-1. Currently, lead stapled peptides targeting this protein are in the preclinical research stage.
In addition to the aforementioned stapled peptides that are nearing clinical application, the growing interest in stapled peptides as therapeutic agents has led to the identification of various lead compounds that regulate disease-associated protein-protein interactions (PPIs). Table 1 summarizes the currently reported stapled peptides with confirmed target interactions and significant pharmacological activity.
Before the advent of stapled peptide strategies using ring-closing metathesis (RCM) to stabilize α-helices, various methods had been reported to stabilize peptide α-helical conformations through side-chain cross-linking, such as amide bonds, azobenzene, hydrazones, and disulfide bonds. However, these modified peptides could not be classified as stapled peptides, primarily due to their inferior pharmacokinetic properties compared to all-hydrocarbon stapled peptides. In recent years, new modification strategies have emerged, where the linking bridges contain heteroatoms yet share the ability to stabilize conformations, enhance activity, and improve pharmacokinetic stability, akin to classical all-hydrocarbon stapled peptides. These methods can be considered generalized stapled peptide modification approaches. Below, they are classified based on the structure of their bridging rings.
The Huisgen 1,3-dipolar cycloaddition reaction in click chemistry enables regioselective formation of 1,4-substituted triazoles via copper-catalyzed reactions between alkynes and azides. Click reactions are highly reactive, selective, and biocompatible, and their cycloaddition products offer stability against proteolytic enzymes. Researchers were among the first to apply this cycloaddition reaction to peptide side-chain cross-linking, achieving satisfactory results. In this method, non-natural amino acids bearing terminal alkyne and azide groups were introduced at the i and i+4 positions of the peptide chain. The purified peptide underwent click reaction in solution, catalyzed by CuSO4 and ascorbic acid, forming a "triazole bridge." The click-modified PTHrP peptide exhibited 5–10 times higher activity than its native counterpart and demonstrated similar bioactivity to amide-bridged PTHrP peptides.
Subsequent studies applied click chemistry to construct triazole bridges in the design and synthesis of BCL-9 stapled peptides. By screening insertion sites and optimizing triazole bridge length, ideal stapled peptides were obtained. Notably, introducing dual triazole bridges into the peptide chain increased its α-helical content to over 90%, reaching a maximum of 99%, while significantly improving its affinity and metabolic stability compared to wild-type BCL-9.
Compared to all-hydrocarbon stapled peptides, triazole stapled peptides benefit from inexpensive and low-toxicity copper catalysts, making them a highly feasible stapled peptide construction method. However, further research is needed to fully understand their impact on α-helicity and enzymatic stability.
In peptide and protein modification, cysteine derivatization has become an essential approach and a key method in chemical biology research. While various cysteine modification techniques exist, including alkylation, conjugate addition, oxidation, and reduction, most peptide stabilization strategies have relied on disulfide bond formation through side-chain thiol coupling. However, disulfide bonds are inherently unstable under oxidative or reductive conditions. By reacting the thiol groups with metabolically stable linkers to form bridges, more stable constructs can be achieved, leading to the development of novel stapled peptide construction strategies.
Researchers have reported using 4,4'-dibromomethyl biphenyl (Bph) or 6,6'-dibromomethyl-2,2'-bipyridine (Bpy) as linkers to form thioether methylene biphenyl or bipyridine bridges via reaction with cysteine side chains at i, i+7 positions. This modification strategy was later applied to a peptide sequence (LTFEHYWAQLTS) with dual inhibitory effects on p53-MDM2/MDMX interactions. Since this sequence does not contain cysteine, Glu4 and Thr11, which are solvent-exposed, were substituted with cysteine to enable bridge formation. Activity screening revealed that the modified stapled peptide exhibited improved α-helical content, enhanced bioactivity, and significantly increased membrane permeability.
The same strategy was used to modify a CAI protein fragment (ITFEDLLDYYGP) reported to inhibit HIV-1 capsid assembly. By replacing solvent-exposed Glu4 and Gly11 with cysteine, bridge formation was achieved. Activity assays demonstrated a significant improvement in membrane permeability, and cell-based viral infection experiments confirmed high inhibitory activity. Notably, one modified stapled peptide (ISF[CELLDYYC]ESGS) exhibited dual inhibition by binding to both the C-terminal domain of HIV-1 capsid protein (KD = 17.5 μmol·L-1) and gp120 protein (KD = 7.4 μmol·L-1), indicating its potential to block both capsid assembly and viral entry.
The peptide NoxaB-(75−93)-C75A (AAQLLRIGDKVNLRQKLLN) from the BH3 protein selectively binds to McL-1, inhibiting its function. To optimize Noxa BH3 activity, researchers applied the thioether methylene biphenyl bridging strategy, substituting Gln77 and Lys84 with cysteine (AACLLRIGDCVNLRQKLLN) before constructing the staple. Biological activity tests on the modified peptides showed substantial improvements in α-helical content, membrane permeability, and enzymatic stability, as well as strong inhibitory effects on MCL-1-overexpressing U937 cells.
A study utilized perfluorophenyl or perfluorobiphenyl linkers to stereospecifically cross-link thiol side chains at i, i+4 positions, forming a thioether fluorophenyl bridge. This method was applied to the modification of the HIV-1 capsid CAI protein fragment. The resulting stapled peptide effectively inhibited HIV-1 formation and demonstrated superior binding affinity, membrane permeability, and enzymatic stability compared to the unmodified peptide. This approach complements the olefin metathesis strategy, as it requires metal catalysts but avoids the use of expensive non-natural alkenyl amino acids, significantly reducing synthesis costs.
In the modification of cysteine side chains, a study employed 3,6-dichloro-1,2,4,5-tetrazine to react with thiols, forming a thioether tetrazine bridge that stabilized the α-helical conformation. Under photochemical conditions, the thioether tetrazine bridge can be cleaved to generate a thiocyanate side chain, which can subsequently regenerate the original peptide in the presence of cysteine.
A study reported the use of thiol–alkene click reactions for stapled peptide and macrocyclic peptide construction. Dienes of varying lengths reacted with two thiol groups from the peptide chain under 365 nm light at room temperature, forming thioether bridges at i, i+4 and i, i+7 positions while exhibiting excellent tolerance to other functional groups. This allowed the reaction to proceed without the need for amino acid side-chain protection.
A comparison of the α-helical content of an Axin stapled peptide formed via RCM and one modified through thiol–alkene click chemistry showed nearly identical circular dichroism profiles. Additionally, a p53 stapled peptide analog constructed using thioether bridges exhibited similar inhibitory activity against the p53-MDMX interaction as its all-hydrocarbon counterpart and selectively induced apoptosis in wild-type p53 cells. This method can be directly applied to unprotected peptides without requiring metal catalysts, and the use of hydrocarbon-like linkers minimizes non-specific interactions with bulky aromatic groups.
A study reported a light-initiated, single-component thiol–alkyne click reaction for efficiently constructing thioether–alkene bridges on unprotected α-helical peptides. Pentynylglycine (M5) and cysteine were introduced at i, i+4 positions, and bridge formation was completed under 365 nm light initiation. This strategy was applied to α-helical peptides regulating estrogen receptor–coactivator interactions. Comparison with all-hydrocarbon stapled peptides revealed that thioether–alkene stapled peptides exhibited substantial changes in lipophilicity and lower cytotoxicity. The presence of a sulfur–carbon double bond allows further functionalization, expanding modification strategies for stapled peptides and integrating traditional thiol–alkyne radical addition into chemical biology research.
A study reported the orthogonal combination of olefin metathesis (RCM) and alkyne metathesis (RCAM) for peptide structural modification. The key step involved introducing two alkenyl amino acids (S5) at i, i+3 and i, i+4 positions, while incorporating non-natural alkynyl amino acids at i, i+7 positions. Due to the orthogonality between RCM (ruthenium-catalyzed) and RCAM (molybdenum-catalyzed) conditions, this method allowed selective control over cyclization sites, enabling precise control of cyclic peptide topology compared to dual-stapled peptides.
This approach was applied to the modification of cyclic peptides targeting GTPase inhibitors, resulting in the development of an improved inhibitor, StRIP3. Although the modified lead compound was not originally an α-helical peptide, this study provides valuable insights for extending classical stapled peptide methodologies.
Stapled peptides enhance the conformational stability of α-helical peptides through side-chain cyclization, significantly improving their affinity for target proteins, metabolic stability, and membrane permeability. By addressing the limitations of both small-molecule drugs and monoclonal antibodies in targeting protein–protein interactions, stapled peptides have emerged as an effective approach for developing drug leads against these interactions.
Analysis of existing studies reveals two main research directions in stapled peptide development. The first focuses on modifying bioactive α-helical peptides—identified through molecular and structural biology research as regulators of protein–protein interactions—using classical all-hydrocarbon stapling strategies. This approach, which was established early, is now advancing towards the first stapled peptide drug approval, demonstrating the feasibility of stapled peptide drug development. The second direction involves synthetic methodology, where researchers employ existing or novel coupling reactions to construct stapled peptides with new structural features. This area is increasingly becoming a focal point at the intersection of organic chemistry and chemical biology. However, since the discovery of bioactive helical peptides largely depends on structural biology (such as crystal structures) or molecular biology findings, research groups focusing solely on synthetic methodologies often use previously reported bioactive stapled peptides as lead compounds or model helical peptides without biological activity as study subjects.
Stapled peptides have become a major research focus in peptide drug development in recent years. As a leading figure in this field has stated, the revolutionary improvements brought by stapled peptides in peptide properties will lead to a resurgence in peptide drug research. It is foreseeable that with continued advancements in stapled peptide research, new findings in structural design, synthetic methodologies, and novel target discovery will emerge. These developments will undoubtedly provide new strategies and solutions for drug research in areas such as oncology, antiviral therapy, antibacterial treatments, endocrine regulation, and diagnostic reagents.
References